Hello all,
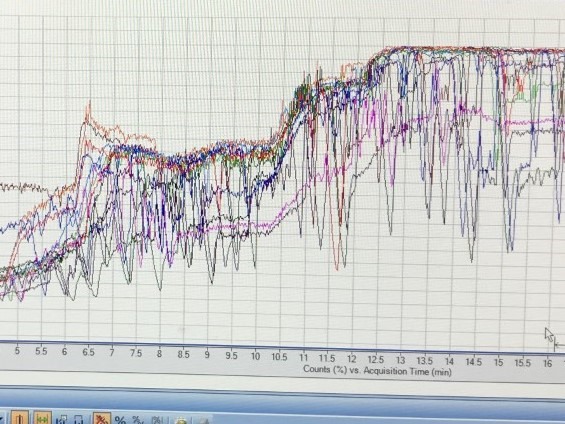
We have having problems with our LC/MS with the TIC signal dropping below the baseline at various points during many runs (see image), including in "blank" samples/washes (both positive and negative mode).
Our setup: 1290 Infinity II LC, diamond hydride silica HPLC column (solvents H2O and acetonitrile, 0.2% acetic acid), with 6545 QTOF (detecting m/z range 50-1200).
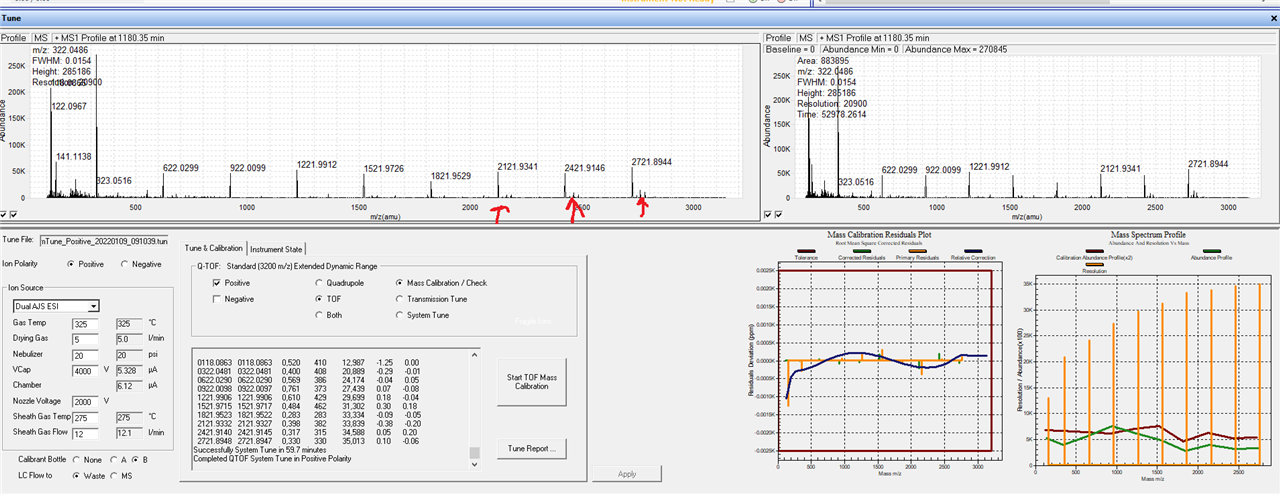
We have tried a new column, different batches of solvent, and re-tuning but no luck...
Help??
(Please excuse my inexperience, I am new to this world).
Thanks,
Will