Dear forum,
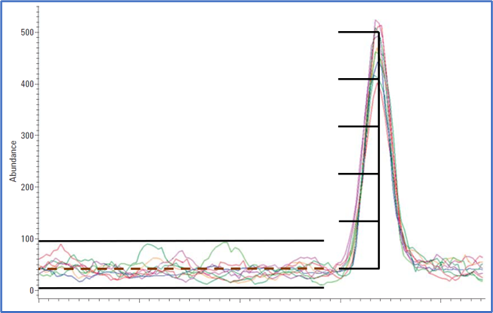
I am attempting to perform untargeted GCMS on plant extracts from ~20 different resurrection plants. My issue is that one peak, namely sucrose, is many orders of magnitude larger than the others in almost all species which is typical for such plants under stress. I extracted from 15mg dry plant tissue with 1 ml 80% MeOH and dried down 300 ul of this extract which produced an overloaded sucrose peak in most species with split 9:1 and gain factor 2. When I dry down 100 ul and increase split ratio to 25:1 it is still overloaded in some species. I also tried lowering gain factor to 1 only over the time interval of the sucrose peak but its still overloaded so I need to load a lot less. However, I am worried that loading even less will result in loosing too many other small peaks of interest as in some of the species the first dilution already lost some interesting low abundant analytes.
My questions concern using the gain factor can be used to deal with this issue.
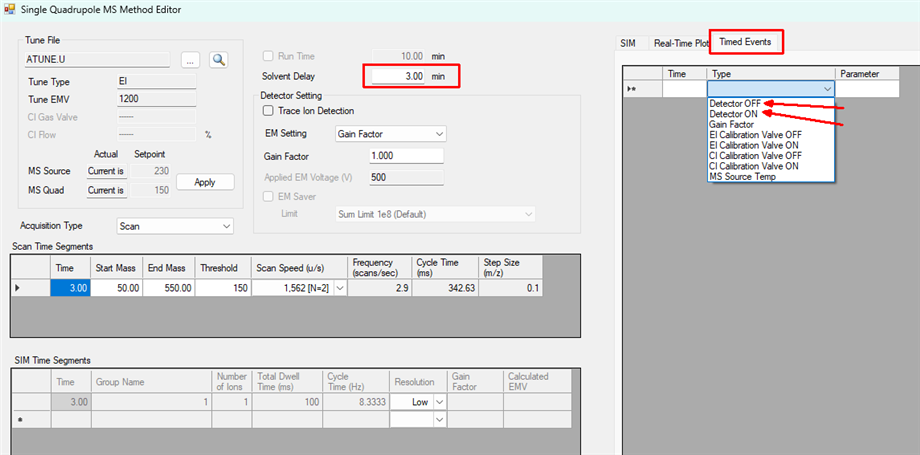
Firstly, will lowering the gain factor at the sucrose peak even further help prevent overloading? My understanding is that if the detector or even column etc is overloaded then changing the gain factor should not make a difference as it would just lower the peak signal but change the flat top of the peak? However, I have seen it mentioned that it does affect overloading in some references.
Alternatively, I am thinking to load even less such that sucrose is no longer overloaded but then substantially increase the gain factor for the retention times around the sucrose peak to boost the signal of the other peaks and so potentially avoid loosing as many. Not sure how well this would work though.
Any advice on how to maximize peak detection, would be appreciated.
P.S. other people have overcome this problem by running each sample twice at a higher and lower dilution but this is not in budget for me as there are 500 samples.
Thanks in advance,
Michael
Instrument: GC Agilent 7890A