I am using a splitless method to analyze a solution of a material that contains non-volatile polymer and volatile monomer I am running a sequence in which use one method to volatalize and analyze the monomers and a few high boiling solvents followed by a second method that degrades the polymer and removes it. I can see that method 2 is pretty effective in cleaning out the liner from the polymer and resulting contamination. We tuned the method untile we had very nice overlaying chromatograms and a very good calibration result.
Then I replaced the filament, the guard column and the MS transferline (both fused silica, deactivated) and all the the connections and ferrules from these to the MS, the PUU (which is used to connect to the analytical column on both sides) and the inlet. After that the calibrations still look fine (area ratios of standard to volatile components), but the overlays don't look good anymore. I even observe shifts in retention time.
When I use split methods for other materials, the overlays look much better.
There is no leak based on the tune and the tune looks very good in general.
I replaced the liner+O-ring, septum and syringe step by step. Still the same issue.
I also just recently replaced the vent line filters.
A consultant of ours suspects the vent valve to maybe not seal properly. Also, I was wondering if it could be the gold seal that needs to be replaced.
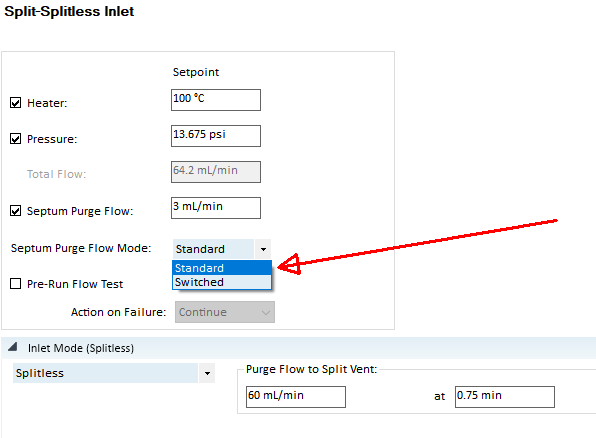
Also it would be great if someone could explain to me a bit more how the sample is injected and volatalized and then released from the inlet in a split/splitless mode. How long is the material heated and not released in the inlet, How long does the vent valve open before it closes again? How far does the syringe go into the liner? Is there a part of the liner that is not heated as much or is the whole liner heated homogenously in the inlet?
My system is an Agilent GC 6890N - MS 5975C.
I used both of these liners: https://www.agilent.com/store/productDetail.jsp?catalogId=210-4004-5, https://www.agilent.com/store/productDetail.jsp?catalogId=210-4022-5 I know they say split, but I was told they both work for splitless as well and since I have some split and some splitless methods that worked pretty well for us.
Thanks. Bettina