We would like to establish a workflow for the semi-quantification of unkowns in our samples.
To do so, we measure a calibration row of a bunch of external standards and our sample. Internal standard is added by co-injection directly in the multisampler.
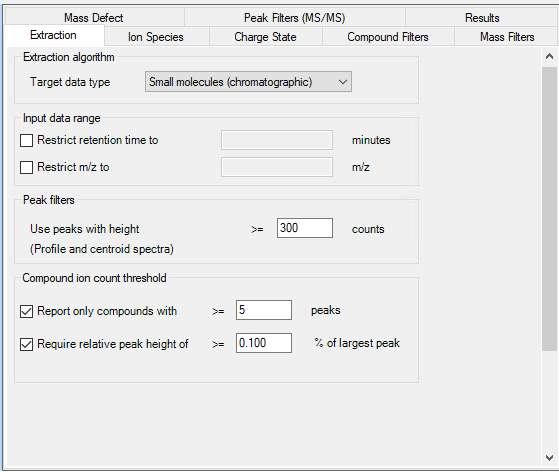
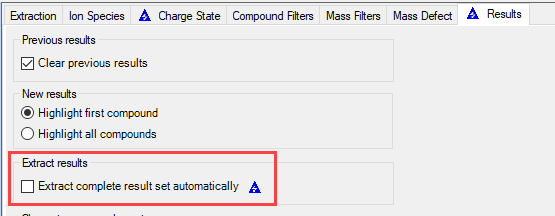
After being measured, we use MH Qual to identify (unknown) compounds in our samples. To do so, we use the FBMF workflow.
When we tested the workflow with our lowest calibration standard (10 ppb), we encountered a problem:
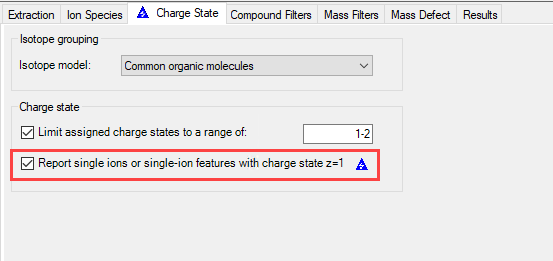
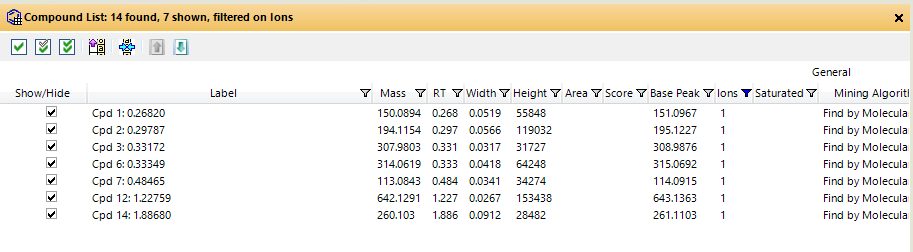
Even though we tried to adjust the quality score and all other parameters in several different combinations, our calibration substance is not recognized as a feature. In contrast to that, when we extract an EIC, we see a nice peak of the substance.
Now my question:
Given the low LOQ, which we cannot increase, is it somehow possible to improve the FBMF workflow to find this feature? Or how else would you tackle this?
We are also reluctant to use the FBF workflow, since we want to do a non-target screening.
After the Qual workflow, we would the import the data (via .cef) into Quant to do a semi-quantification.
We use an Agilent QTOF 6546
Software: MassHunter Qualitative Analysis Version 10.0 Service Release 1