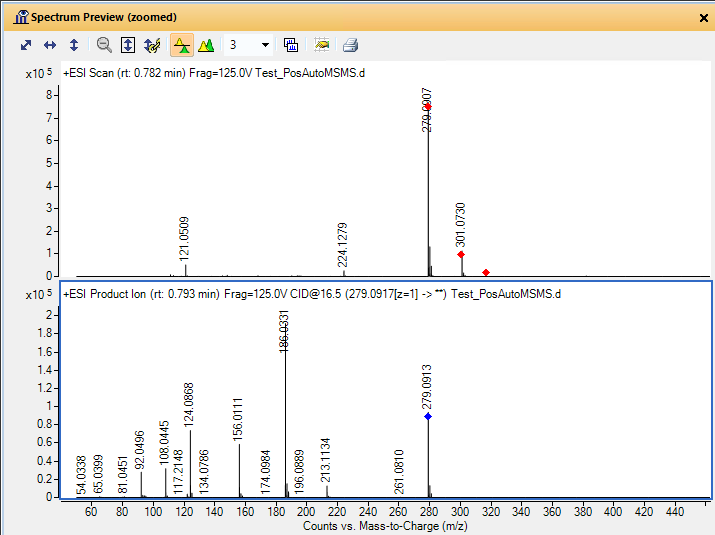
I am a new intern at a dry lab looking at mass-spec data from our wetlab partner. The wet lab is doing DDA using an Agilent 6550 Q-TOF to analyze some biosamples. When looking at the output .mzml file, I am noticing something unexpected in the m/z values of the precursors and their associated fragments. For example, in observation #1, the precursor m/z is 535.772527 with a charge of two. The fragments array associated with this precursor has 331 fragments. The sum of the m/z of those fragments is 165,958.62561798096. My understanding is that using DDA, a precursor is identified and then broken down into its fragments. If so, how can the sum of the m/z of the fragments be so much larger than the precursor m/z? If the fragments are not independent, how does the mass spec make multiple reads of the same fragment? Is there actually multiple precursors with the exact same m/z value generating all of those fragments?
Thanks in advance.