

Hello,
Disclaimer: I am new to the software. I have created a processing method with over 100 potential compounds I want to look for. My samples (known mass) are spiked with a known mass of standard solution containing 3 standards in different concentrations (ppm). The point I am stuck at the moment is is that after setting my samples to spiked, I cannot get the identified peaks to be quantified, and I also have some minor questions regarding the processing method as well. Details are as follows:
Software: OpenLAbs CDS v 2.7
Equipment is a GC-FID/MS to simultaneously quantify and qualify a sample.
What the issue is:
- I only see an amount and concentration in the results tab for the 3 standards I've inputted.
- The calibration curve table or internal standard peaks says "Peak is ISTD", no calibration data abailable
- For other compounds, the calibration table shows "not quantified", with also no calibration data available.
What I've done so far:
- In the injection list, I've set all samples to "spiked". I've added a sample amount (g) to each. ISTD amount 1-3 are blank atm.
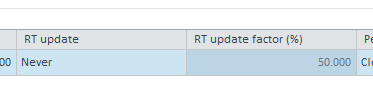
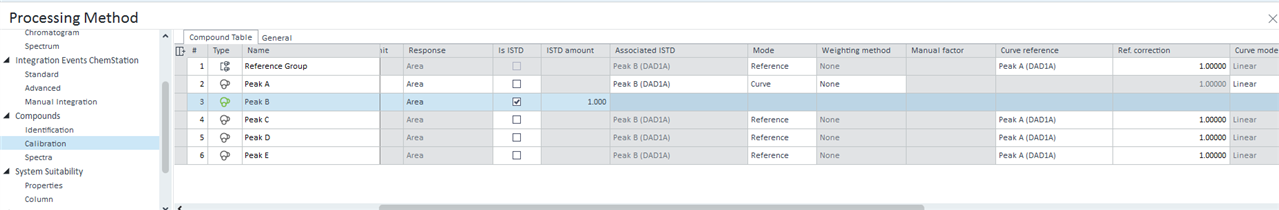
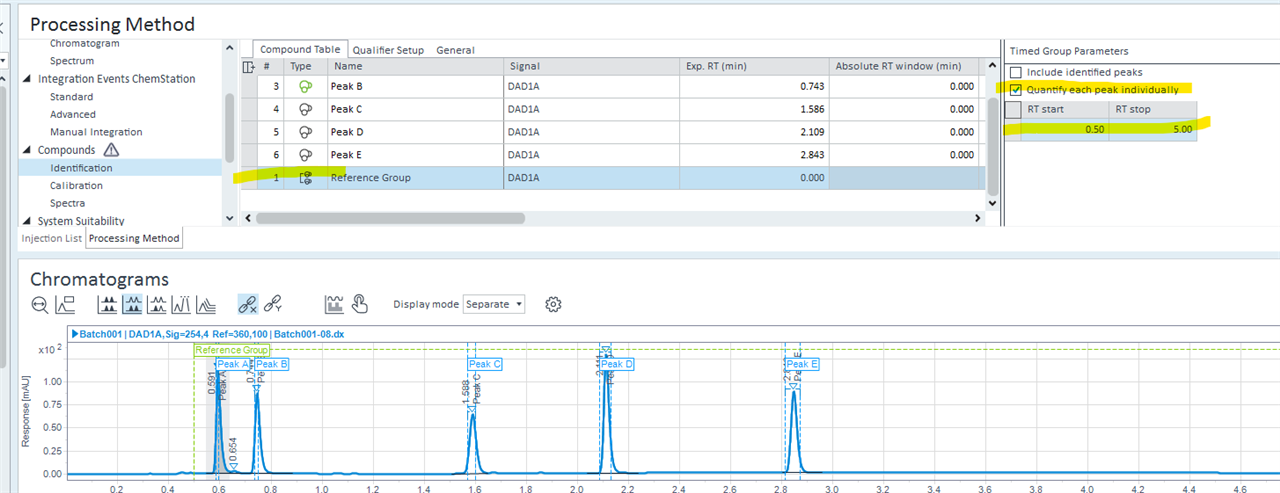
- In the compounds indetifiaction tab, I've assigned my 3 standrads as standards in the compound list and assiciated all the remaining compounds to either of them as associated time reference compunds.. RT update after each run for every comound.
- In the compound table of the Compounds calibration tab, I have is ISTD selected for my 3 standards, with their concentrations in PPM inputted to ISTD amount. I have assigned all the other compounds to either standards. Mode: curve, Weighing method: none, curve model: linear, Origin: force, response scaling: none.
- In the general section of the compounds calibration tab, I have internal standard selected. Number of levels: 1. Curve calculation: from average per level. RF definition: Amount per response.
Additionally, a question about the processing method: out of my 100 potential compounds, some do not occur in all the samples. i've set every compound to adjust for retention time shift based on the shift of my internal compounds in the method. Is there a way to also shift the expected retention times of undetected compounds in my method?
Also, is there a way to automatically identify unidentified peaks in the FID spectra using the MS spectra? As in if a peak is not identified, the program would use the MS data to provide the most likely match?
Thank you in advance for the help.