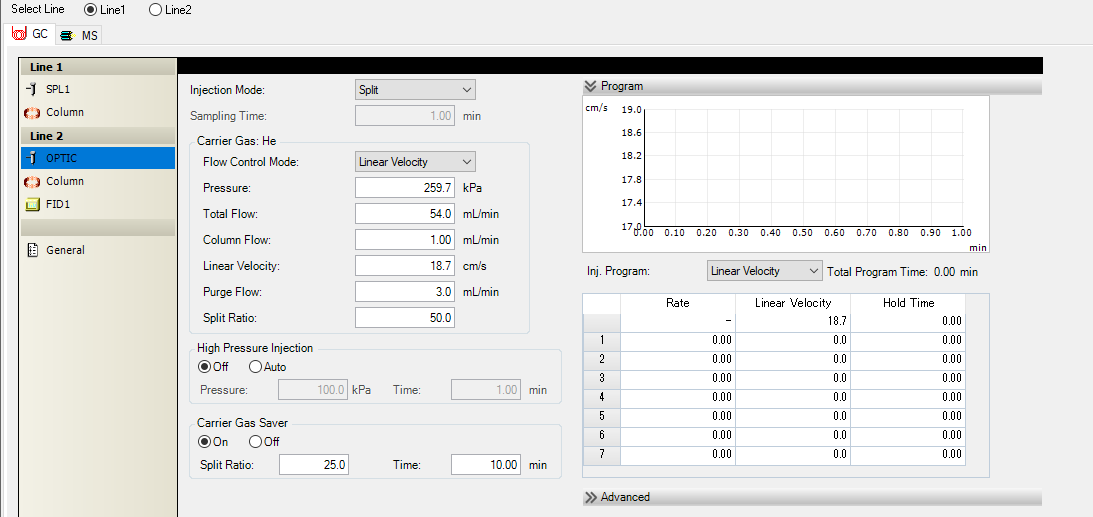
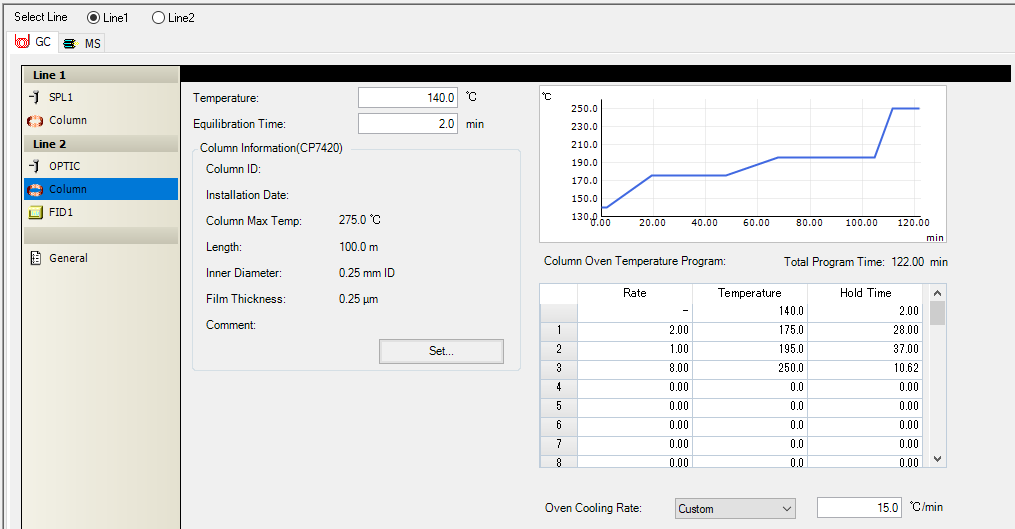
Hello, I have a Agilient CP7420 column connected to the GCMS-QP2020NX also equipped with a FID detector and the column is connected to the FID (carrier gas helium 1mm/min). The column and the chromatograph are new, but the column was purchased from 3 years ago but was not unpacked
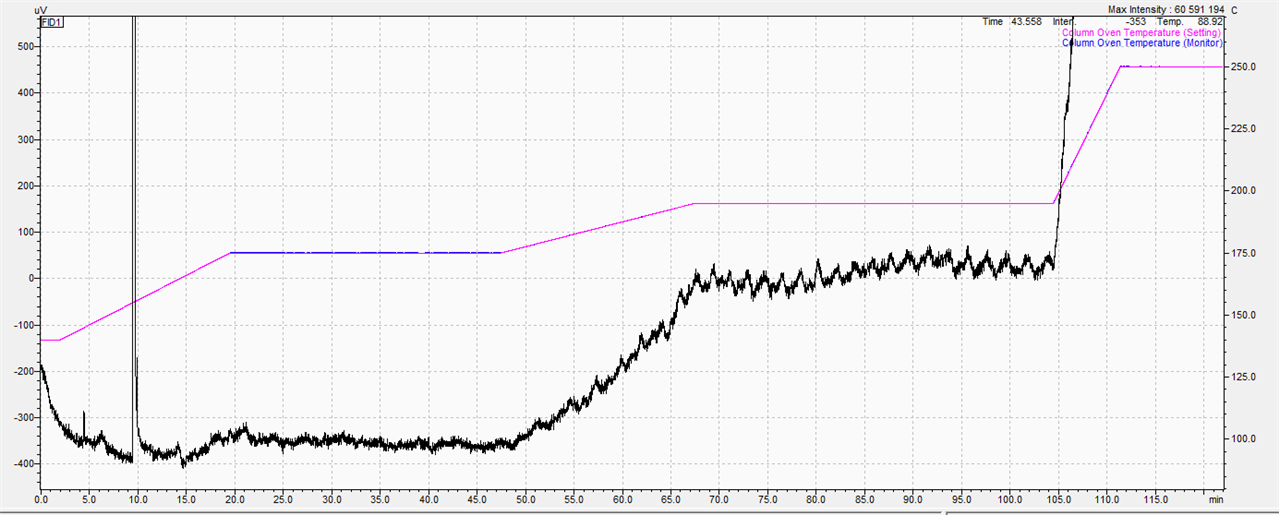
In terms of FAME separation, I am very pleased with the column. However, I have some problems with an unstable line at higher temperatures which generally make it very difficult and sometimes impossible to quantify the analytes.
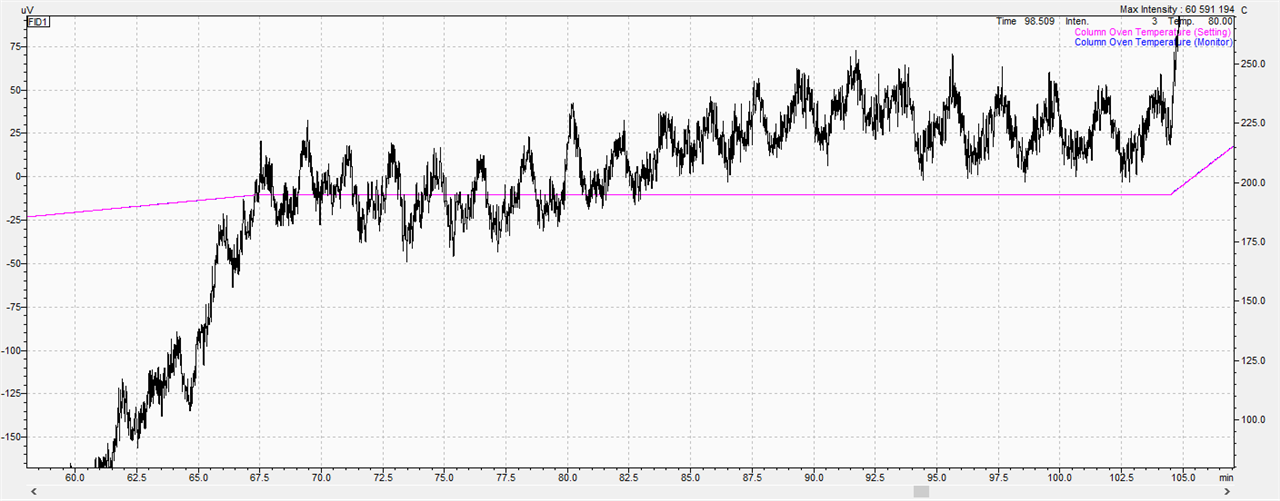
Above 175C, irregularities begin which, with the appropriate enlargement, look like 'saw blades'.
Basically, all procedures related to the chromatograph were performed (column conditioning for many hours at different temperatures, cleaning of the injector and FID, the split filter was replaced, the silica gel in the hydrogen generator was dried, the septa and liner was replaced, the column was cut several times. and reinstallation, filters are installed on both the carrier gas and the detector gas lines).
The manufacturer's service after performing all the above-mentioned steps found that these instabilities came from the column. My question is whether it is possible to deal with the fluctuations attached in the picture or Is it just the column's fault? Or maybe the injections of various solvents, such as methanol or acetone, can help. so far, n-hexane has been used
I would be grateful for any help and suggestions