

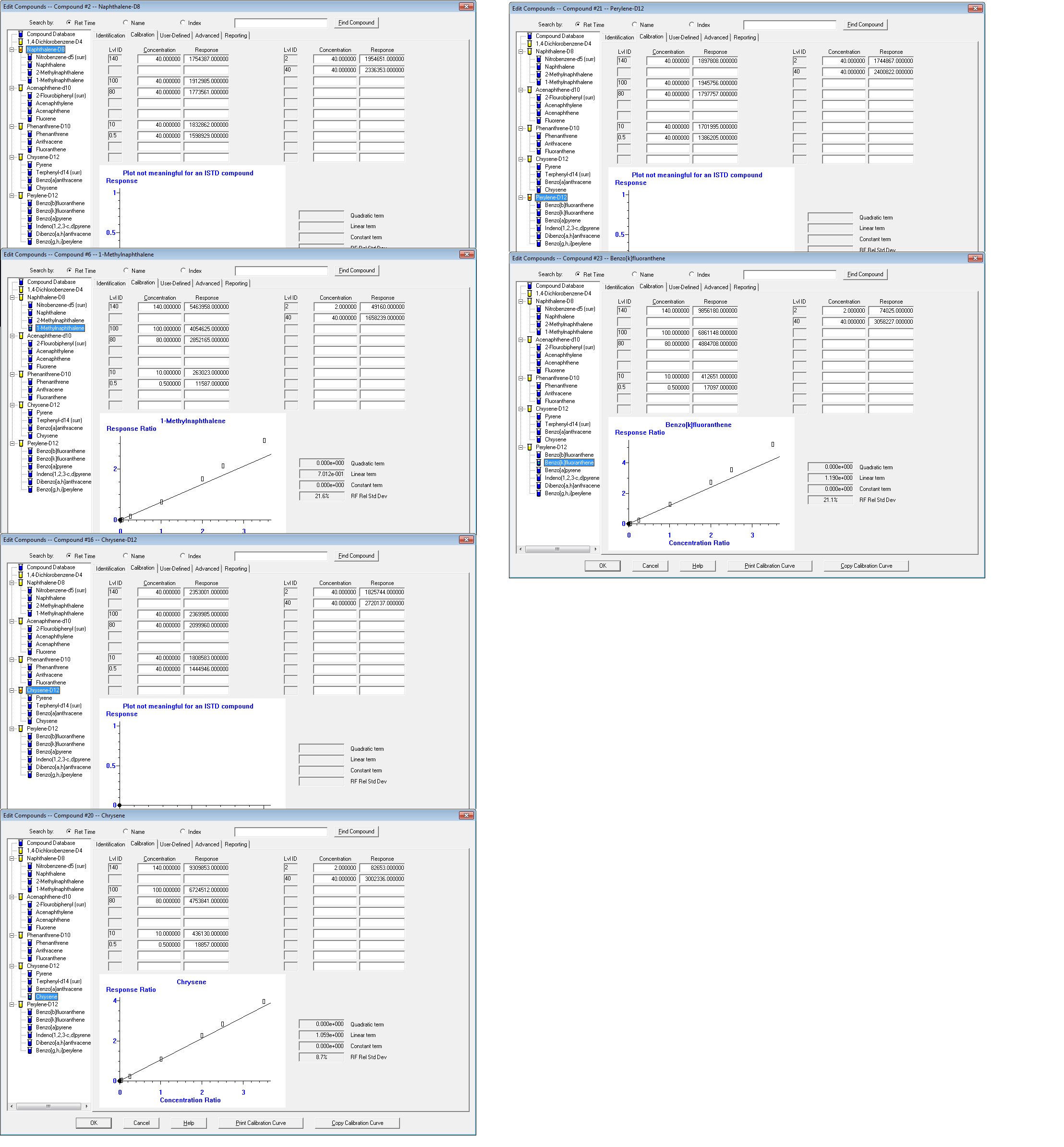
I please ask if you can verify what I am doing wrong. My analysis is 8270 PAHs using a 7890A/5975C inert XL MSD. I currently use 30m x 0.250mm x 1.00µm DB-5MSUI (Part # 122-5533UI) Agilent column. The attachment below shows my RSD% for three of my four groups of analytes over 15% with noticeable peak deviation in the final CC concentrations. I have made the calibration curve with the desired concentrations more than once with the same instrument results. I will greatly appreciate your help.
Current column settings:
1) 50°C (1min)
2) 15 °C/min to 300° - hold 4.5min
3) 25 °C/min to 320° - hold 12.5
Total time 35.467min
Current GC settings:
MS Source 230°C, MS Quad 150°C, Aux Temp 310°C, HiVac 1.14e-05
Inlet: Temp=280°C, Pressure=10.052psi, Flow=24.20mL/min, Constant Flow
Splitless injection at 50mL/min at 1min



{kind=link}