

I'm working on developing a suite of in vitro ADME assays (specifically forced degradation right now) most of which center on using HPLC-MS. The assays themselves are well developed and I can run them efficiently but I'm facing a serious bottleneck at the data processing stage.
I run these assays on an Agilent Infinity 1290, processing with Agilent CDS Chemstation Edition. The forced degradation assays typically run like this:
- Set up a sequence where DMSO stocks are diluted with a variety of buffer conditions (e.g., 0.1 N HCl with 20% ACN at 0.1 mg/mL).
- Run the sequence to generate t0 data. The three pieces of data that are collected at this stage are retention time, % area, and m/z of base peak in corresponding ESI positive MS trace. These three pieces of data are associated with a peak that we will track as the assay continues. Collect this data for all peaks with area > some arbitrary value.
- Using the command scheduler, schedule sequences for 2, 4, 8, 16, and 24 hours from t0.
- As time points are taken, populate an excel spreadsheet in a table like the one below.
| m/z | RT | t0 | t2 | t4 | t8 | t16 | t24 |
|---|---|---|---|---|---|---|---|
| 395 | 1.016 | 100 | 100 | 99.1 | 97.5 | 94.9 | 92.3 |
| 162 | 1.095 | 0 | 0 | 0.9 | 1.8 | 3.8 | 5.7 |
| 377 | 1.153 | 0 | 0 | 0 | 0.6 | 1.3 | 1.9 |
Here are the issues I'm encountering in this process:
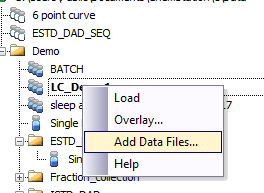
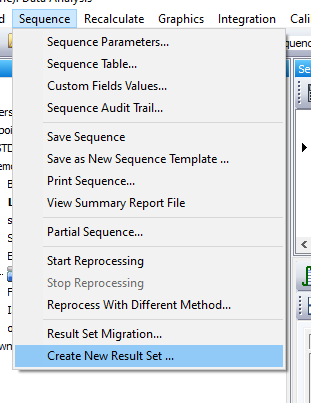
- Each timepoint is run as a new iteration of the forced degradation sequence. Ideally, all timepoints for a given sample would be exported into a new sequence (or some other meta-organization data structure) so that I could easily follow the degradation. Presently, I have to flip back and forth between sequences looking for the same sample name before I can populate the excel spreadsheet. This adds time and requires more focus than I'd like. It is possible to load signals from other sequences into a new result set as I'm learning right now, but I'm still not sure how to do this automatically.
- I have to manually identify and track peaks across timepoints, making judgement calls about whether peaks are real or if they're erroneous. Further, I have to confirm that the peaks in later time points are the same as peaks from earlier timepoints. Ideally, I could use automation to follow peaks over time and populate the table with relevant data.
- I'd like to generate stacked plots of chromatograms across timepoints for a given sample. Chemstation has some tools for this but they're extremely clunky and I'm never fully convinced that I've figured them out even when I make good looking plots.
These are my main thoughts about improving my data processing and analysis. Hopefully someone out there has encountered this situation before and knows how to help.



 ]
]