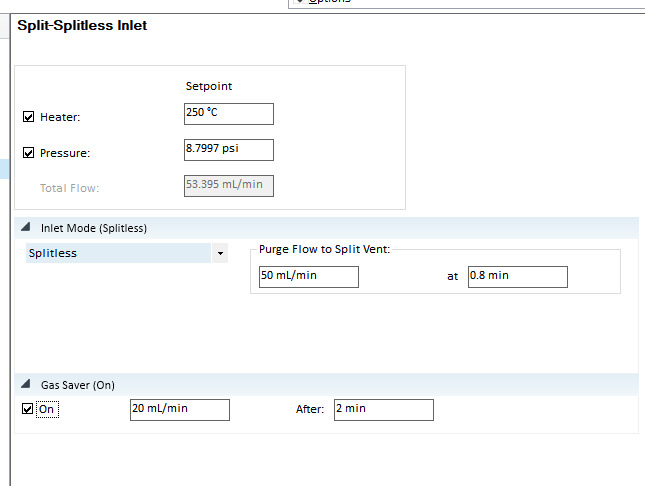
Why is the purge flow affecting the column flow, e.g. we set purge flow to 0.5 mL/min, and that leads to the column flow of 1.4 mL/min not being reached.
Why is the purge flow affecting the column flow, e.g. we set purge flow to 0.5 mL/min, and that leads to the column flow of 1.4 mL/min not being reached.